


- Highly accurate 'chain termination based' 'first generation' DNA sequencing method
- Gold standard for benchmarking all other sequencing technologies, including NGS
Sanger

Sanger Sequencing
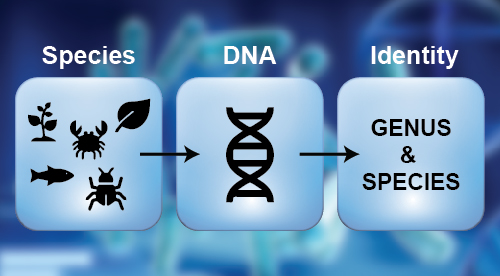
Identification of Organisms (Isolate-to-Identity)
- PCR amplification with universal gene primers.
- Cycle sequencing using BDT v3.1 chemistry followed by sequence assembly and BLAST in relevant database

Regulatory / NABL compliant Services
- Services are accorded as per NABL compliance ensuring the best quality
- Shortest turnaround time
- Reports formats customized to suit your regulatory requirements

Supplemental Services
Only offerred in combination with other Sanger service codes, NOT separately
×
Sanger Sequencing Applications
- Highly accurate 'chain termination based' 'first generation' DNA sequencing method
- Gold standard for benchmarking all other sequencing technologies, including NGS
- Confirm sequence variants or fill ‘gaps’ of genomic regions determined by NGS
- Predesigned identification service under Isolate-to-Identity covering all organisms
- Detailed reports always include .ab1 files and other useful information
- Standard vector primers and universal gene markers added free-of-cost
×
Supplemental Services
Only offerred in combination with other Sanger service codes, NOT separately
- Agarose Gel Extraction
- Plasmid DNA Isolation
- Purification of PCR Products
×
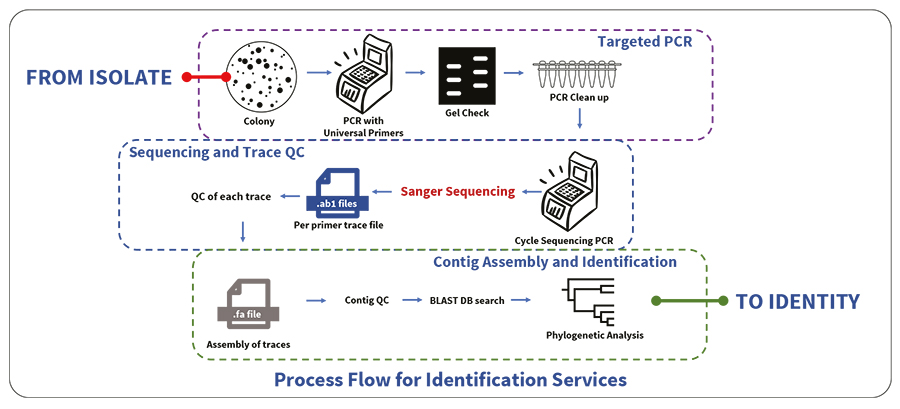
Identification of Organisms (Isolate-to-Identity)
- PCR amplification with universal gene primers.
- Cycle sequencing using BDT v3.1 chemistry followed by sequence assembly and BLAST in relevant database
- Phylogenetic analysis (MBS020) is included in all identification services, except where indicated.
×
Regulatory / NABL compliant Services
- Services are accorded as per NABL compliance ensuring the best quality
- Shortest turnaround time
- Reports formats customized to suit your regulatory requirements
NGS (Next Generation Sequencing)

Whole Genome Sequencing (WGS) Applications
Whole Genome Sequencing (WGS) has enabled comprehensive decoding of the sequence of entire genomes of organisms. Such information is crucial for understanding functional and evolutionary history of organisms...

Whole Exome Sequencing (WES) Applications
The human genome has 180,000 coding regions which account for 1.7% of the genome. An estimated 85% of human diseases with genetic basis occur due to aberrations in these coding regions.

Amplicon-Based Microbiome Sequencing Applications
- A potential tool for high-throughput phylogenetic analysis of microbial communities.
- Based on phylogenetic marker genes; 16S, 18S rRNA genes & ITS region

Shotgun-based Metagenome Sequencing Applications
Shotgun metagenomics is a proven tool to study environmental, agricultural & human microbiomes,
pathogen identification & surveillance as well as monitoring of anti-microbial resistance genes in the
environment.
Transcriptomics
RNA sequencing (RNA-seq) is a powerful and widely used technique for transcriptome analysis that
has revolutionized the field of molecular biology. It allows researchers to study gene expression at an
unprecedented level of detail, providing valuable insights into the mechanisms of cellular processes...
Ready-to-load / Custom NGS
- User supplies ready-to-run library pool and is responsible for data quality, read distribution or failure due to user errors.
- Sequencing using relevant PE read chemistry to generate data.
- Sequencing Run information.
Supplemental Services
Only offered in combination with other NGS service codes, NOT separately
×
Whole Genome Sequencing Applications
Whole Genome Sequencing (WGS) has enabled comprehensive decoding of the sequence of entire genomes of organisms. Such information is crucial for understanding functional and evolutionary history of organisms,as well as to identify mutations and genetic variations that are responsible for causing genetic disorders,development of cancer, disease outbreaks, evolution of strains with novel pathogenic traits, etc. At Hi-Gx360 ®, we use a read-to-annotation approach to generate in detail all the information needed to understand functions of all predicted genes present in the organism. Assembled genomes may not be 100% complete, hence, further gap-filling can be achieved using one of many long read sequencing technologies.
- Sequence close relatives of reference-organisms or entirely novel organisms (de-novo)
- Obtain high-resolution and accurate genomic characterization using 2 x 150 or 2 x 300 bp reads
- Data supplied with interactive QC report.
- Predesigned identification service under Isolate-to-Identity covering all organisms
- Receive raw sequence reads, assembled contigs & scaffolds Along with bioinformatics analysis customised to fit your needs
- Choose from basic or advanced bioinformatics.
Recommendations
| Resequencing | De-novo Sequencing | |
|---|---|---|
| Depth/Coverage | 50X - 100X | >100X |
| Read length chemistry | 2x75 to 2x300 | |
| Sample Requirements | Minimum quantity 0.5 μg, Minimum concentration = 20 ng/μl, A260/280 = 1.8 - 2.0 | |
Deliverables
- Run information, Statistics of raw data & FASTQ files
- Reference Genome Mapping files/ De-novo Genome Mapping files, Functional Annotations, Gene predictions,Gene annotations, Pangenome analysis
- Advanced analysis customized to your project needs
×
Whole Exome Sequencing Applications
The human genome has 180,000 coding regions which account for 1.7% of the genome. An estimated 85% of human diseases with genetic basis occur due to aberrations in these coding regions. Targeted sequencing of these regions of the genome are termed as whole exome sequencing (WES). Exome sequencing is a more cost-effective option than sequencing the whole human genome.
This sequencing method uses a targeted capture approach where biotinylated probes are deployed against the coding regions of the fragmented human genome to capture and pull down these parts with streptavidin coated beads. The fragments thus obtained are sequenced on the NextSeq™ 550 platform. This strategy results in a 100-fold increase in coverage of the relevant sections of the genome.
- Lower cost and wide availability
- Sequencing coverage on target upto 30X
- Detection of coding single-nucleotide polymorphism (SNP) variants as sensitive as whole genome sequencing
- A smaller data set for faster and easier analysis compared to whole genome sequencing
- Standard sequencing coverage ≥ 50X; cancer sample ≥ 100X. More SNPs can be gained by increasing the coverage.
- Cost-effective library preparation and exome enrichment solutions
Exome Sequencing Recommendations
| Whole Exome Sequencing | Cancer Specific and/or Rare Variant Detection | |
|---|---|---|
| Depth/Coverage | 100-300X | 500–1000X |
| Read length chemistry | 2x75 to 2x150 | |
| Capture Region | Agilent SureSelect Human All Exon V8, Design size 41.6 Mb, Target region 35.1 Mb | |
| Sample Requirements | Required quantity 1.0 – 0.2 μg, Minimum concentration = 20 ng/μl, A260/280 = 1.8 - 2.0 | |
Deliverables
- Run information, Statistics of raw data & FASTQ/VCF files
- Mapping statistics, Statistics of sequencing reads
- SNPs and InDels calling, Variant annotation, SNVs concordance, Tumor-Normal paired analysis
×
* Note: Number of reads may be decided by client and may vary depending on the diversity in the sample
Amplicon-based microbiome sequencing Applications
- A potential tool for high-throughput phylogenetic analysis of microbial communities.
- Based on phylogenetic marker genes; 16S, 18S rRNA genes & ITS region
- Allows species identification and taxonomic diversity characterization
- Provides detail insight of genus and species
Advantages of amplicon-based phylogenetic gene marker sequencing
- Marker genes are omnipresent
- Marker genes are highly abundant to those of other genes
- Measures phylogenetic relationships across different taxa
- Easily estimate relative abundance of microbial communities
- Highly cost-effective
Recommendations
| Objective | Taxonomic profiling |
| Reads per sample | Over 1 Lakh paired end reads* |
| Read length chemistry | 1x150 to 2x300 |
| Sample Requirements | Minimum Quantity 1 μg, Minimum Concentration = 50 ng/μl, OD 260/280=1.8~2.0 |
Deliverables
- Run information, Statistics of raw data & FASTQ files
- Taxonomic profiles, Functional Annotations, Metagenome assembled genomes (MAGs)
- Advanced analysis customized to your project need
×
* Note: Number of reads may be decided by client and may vary depending on the diversity in the sample
Shotgun-based Metagenome Sequencing Applications
Shotgun metagenomics is a proven tool to study environmental, agricultural & human microbiomes, pathogen identification & surveillance as well as monitoring of anti-microbial resistance genes in the environment. Some of the most common applications include:
- Identification and classification of microbial communities
- Novel biomarker discoveries
- De-novo assembly & characterizations of novel genomes
- Discovery of novel enzymes and metabolic pathways
- Relative abundance analysis of microbial communities
Recommendations
| Objective | • Discovery metagenomics • Relative abundance of microbial communities • Metagenome assembled genome sequencing |
| Reads per sample | 25-500 Million paired end reads* |
| Read length chemistry | 1x150 to 2x300 |
| Sample Requirements | Minimum Quantity 1 μg, Minimum Concentration = 50 ng/μl, OD 260/280=1.8~2.0 |
Deliverables
- Run information, Statistics of raw data & FASTQ files
- Taxonomic profiles, Functional Annotations, Metagenome assembled genomes (MAGs)
- Advanced analysis customized to your project need
×
Transcriptomics
RNA sequencing (RNA-seq) is a powerful and widely used technique for transcriptome analysis that has revolutionized the field of molecular biology. It allows researchers to study gene expression at an unprecedented level of detail, providing valuable insights into the mechanisms of cellular processes and disease pathogenesis. RNA-seq works by converting RNA molecules into a library of cDNA fragments that are then sequenced using high-throughput sequencing technologies.
- Identify and quantify gene expression levels, detect novel transcripts & investigate alternative splicing patterns
- Detect post-transcriptional modifications such as RNA editing
- Wide dynamic range and low input requirements
- Work with a variety of sample types, from single cells to complex tissues.
- Integrate with other types of omics data to obtain a more comprehensive view of biological systems.
×
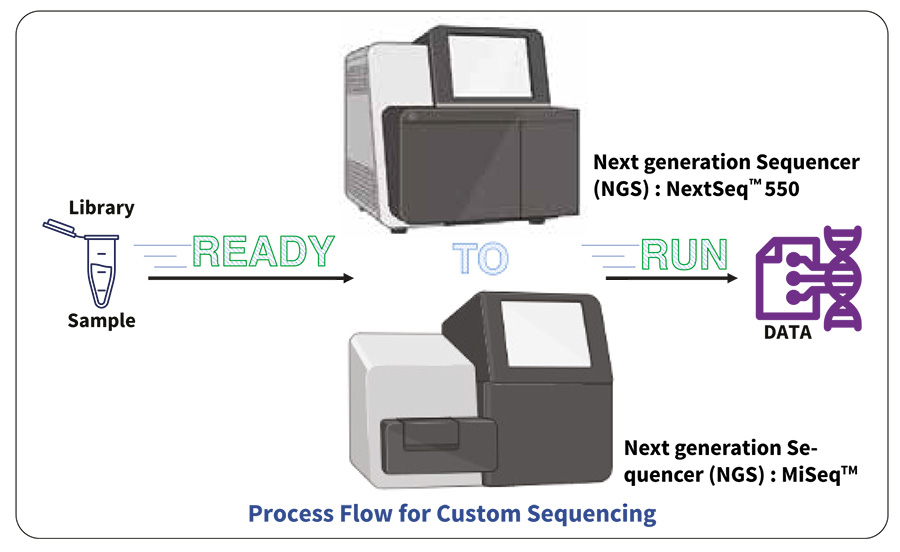
Ready-to-load / Custom NGS Services
- User supplies ready-to-run library pool and is responsible for data quality, read distribution or failure due to user errors.
- Sequencing using relevant PE read chemistry to generate data.
- Sequencing Run information.
- Raw data information (number of quality reads generated including average Q score), adaptor trimmed and clipped reads as fastQ files.
Custom Sequencing
×
Supplemental Services
Only offered in combination with other NGS service codes, NOT separately
Bioinformatics Services
Whether you have smaller sequence datasets or large datasets from NGS, one of the biggest challenges especially in massively parallel sequencing is their analysis. Problems arise mainly due to lack of scalable computational power and lack of knowledge regarding how different bioinformatic (BioIT) tools work under the hood. Data privacy too remains a daunting challenge for researchers who are working with patient data. At Hi-Gx360® we have systematically addressed each of these concerns in our state-of-the- art bioinformatics data center. This includes :
- Secure Datasets that are never shared over the open internet
- Transparent and Open information sharing about analysis workflow.
- Highly Scalable and validated open-source packages used for analysis.
- Secure Interactive reports that are comprehensive and information rich.
- Publication grade data visualizations
- Direct Consultations with our highly qualified scientists on project design

Sanger Data Analysis
- Phylogenetic Analysis (16S/18S/ITS/any other gene based)
- NCBI submissions
- Primer Designing

NGS Data Analysis
- Adapter trimming and reads quality control
- High Quality Denovo assembly of isolates
- Reference guided annotation (assign functions)
×
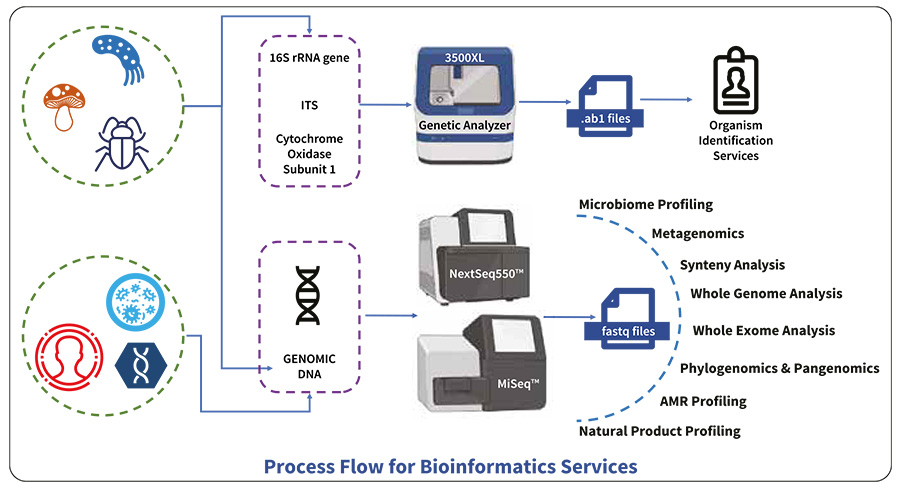
Bioinformatics Services
Whether you have smaller sequence datasets or large datasets from NGS, one of the biggest challenges especially in massively parallel sequencing is their analysis. Problems arise mainly due to lack of scalable computational power and lack of knowledge regarding how different bioinformatic (BioIT) tools work under the hood. Data privacy too remains a daunting challenge for researchers who are working with patient data. At Hi-Gx360® we have systematically addressed each of these concerns in our state-of-the- art bioinformatics data center. This includes :
- Secure Datasets that are never shared over the open internet
- Transparent and Open information sharing about analysis workflow.
- Highly Scalable and validated open-source packages used for analysis.
- Secure Interactive reports that are comprehensive and information rich.
- Publication grade data visualizations
- Direct Consultations with our highly qualified scientists on project design
With our bioinformatics services, you can accurately achieve the following
- Identify organisms with confidence
- Assemble, annotate and analyze whole genomes
- Locate low frequency mutations & identify InDels
- Uncover genetic traits, such as antibiotic resistance markers, etc.
- Combine with short and long reads libraries to obtain complete genome sequence
- Identify repetitive regions, structural variants and complex rearrangements for de-novo assemblies

Natural Product Discovery
Microorganisms are some of the greatest chemists known to man and produce several bioactive chemicals. Genome mining can allow us to predict the biosynthetic potential of the organism even before attempting a costly process of chemical characterization. Such mining of genomes can also inform the chemical synthesis process thus making the process from discovery to production shorter.
MBS010 Genome Mining for Natural Products from Bacteria Archaea (requires MBS001)
Antimicrobial Resistance Profile
Antimicrobial resistance (AMR) is a global threat to human health and
development. In fact, AMR is one of the top 10 global public health
threats facing humanity as declared by WHO. Hence, it is integral to
identify control and prevention strategies to combat the increasing
threat of AMR. Among the many options, genome mining allows for
an accurate prediction of AMR for bacteria. At Hi-Gx360®, our team
will help you identify the AMR markers from the genomic sequence
of your organism.
MBS011 Genome Mining for AMR and other features from Bacteria/Archaea (Requires MBS001)
In-silico Isolation for Mixed Cultures
Sometimes mixed microbial colonies are inadvertently sequenced
together. In such cases all is not lost, it is possible to separate these
genomes bioinformatically. Often this approach offers critical
insights into the two organisms allowing for devising strategies to
isolate them and further analyze them. Genome segregation can also
offer clues into why these organisms grow together thus revealing
mechanisms of cross-feeding based associations.
MBS019 In-silico genome isolation for mixed cultures
Hi-Gx360® Sequencing & Bioinformatics Services#902790
